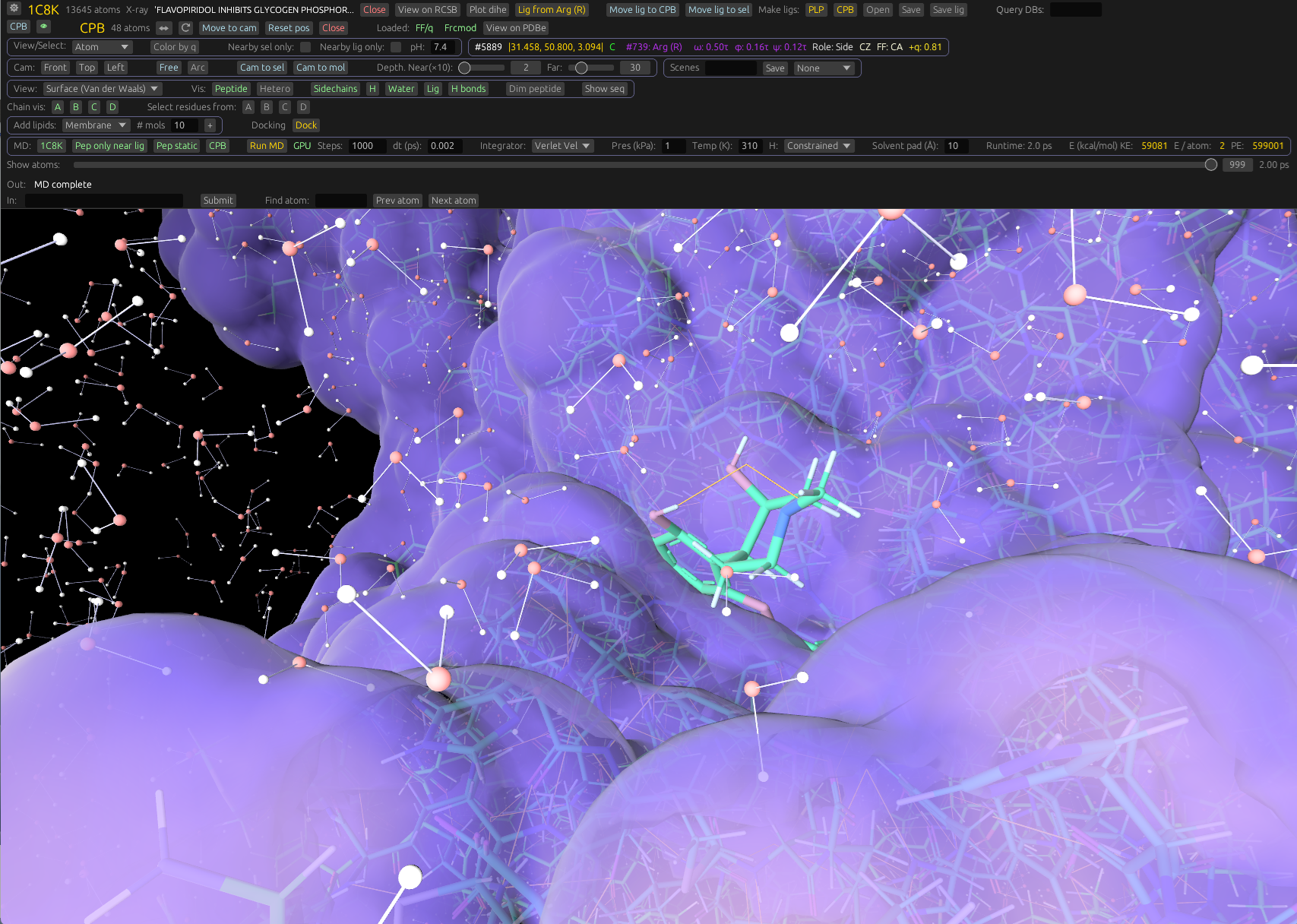
An integrated tool for structural biology
Load from PDB, PubChem and other databases. Run MD with Amber-style parameters, view electron density, and edit small molecules — all in a GPU-accelerated Rust application.
Specialized tools for molecular docking, and simulating liposomes and lipid nanoparticles (LNPs).
Powerful visualization and camera system that lets you explore molecular systems without friction.
Windows and Linux binaries
Why Daedalus
MD built in
Run molecular dynamics with Amber force fields and OPC water directly in the viewer. No separate pipeline.
Density and docking
Render 2Fo-Fc maps, isosurfaces, and visualize docked ligands from crystallography or Cryo-EM data.
Fast and modern
Rust, CUDA, and thread pools for parallel work. Pull structures from RCSB, PubChem, DrugBank, PDBe.
What you can do in minutes
1. Load a structure
Open mmCIF/PDB, or just query a PDB ID. Ligands and parameters can download automatically.
2. Inspect and validate
Switch to surface, stick, or density views; orbit around selections; measure bonds and dihedrals.
3. Run MD
Start a short MD to see realistic conformations of ligands, nucleic acids, or proteins.
Gallery
Representative views from the README
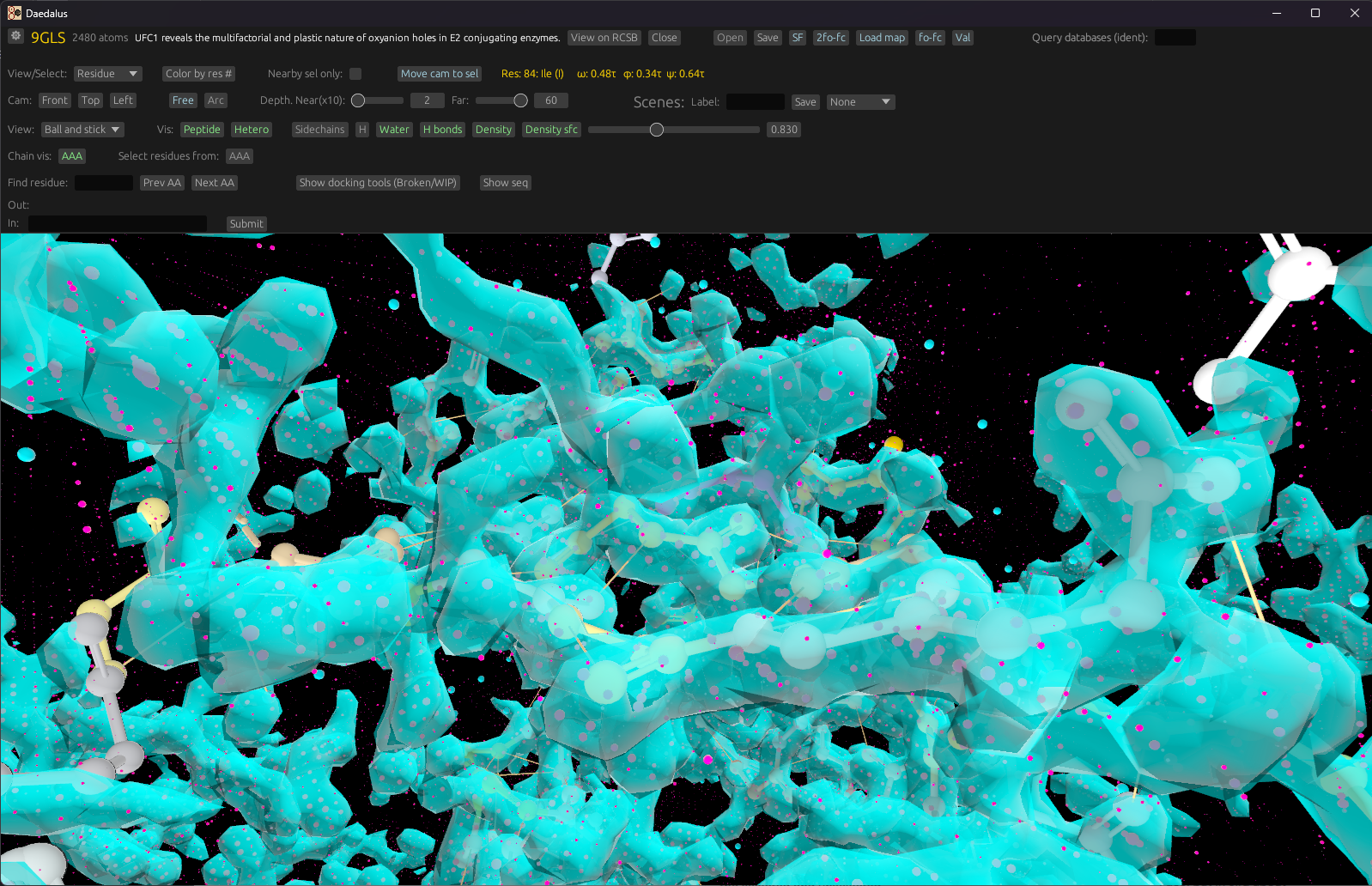
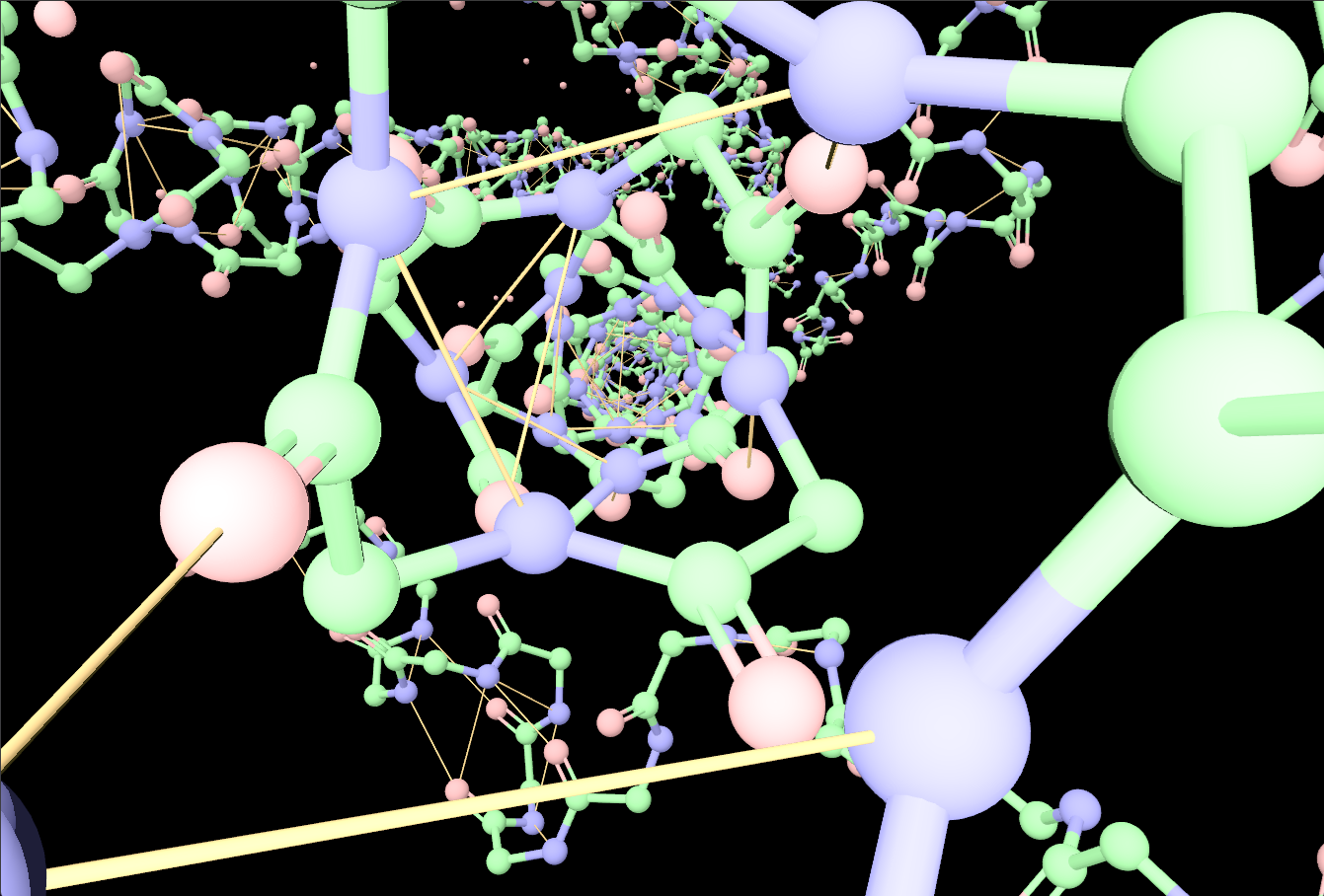
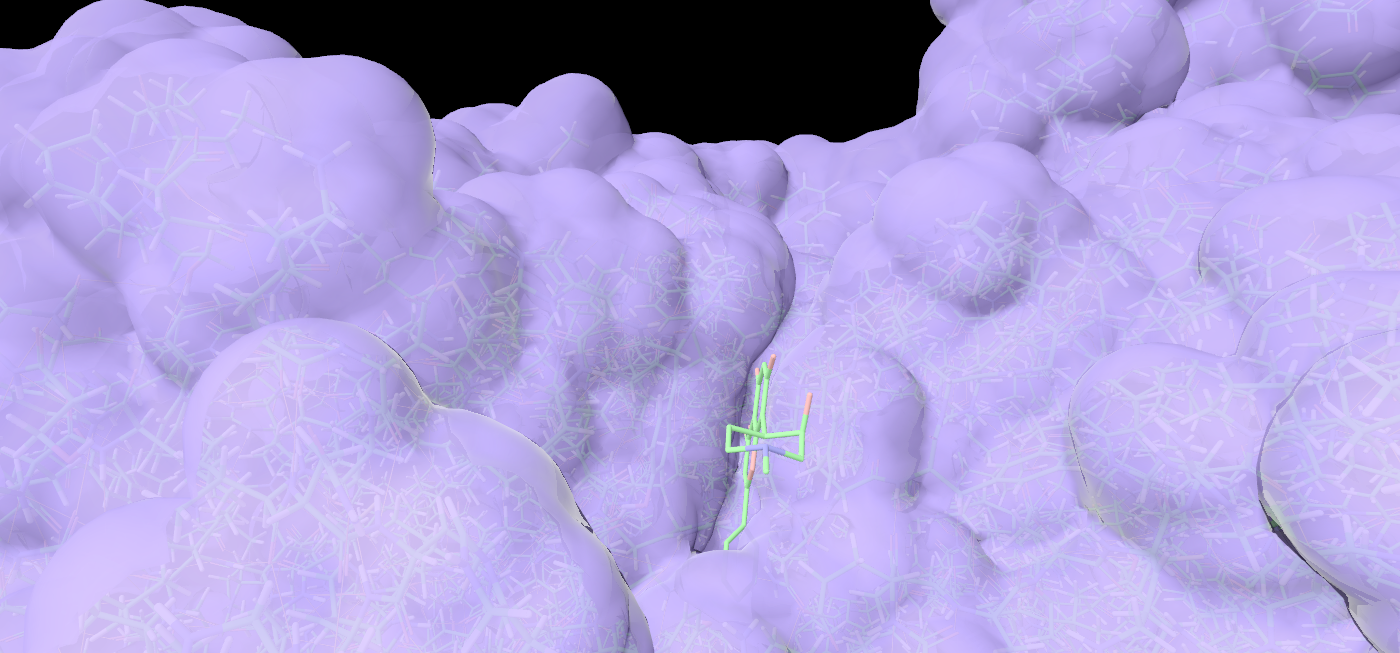
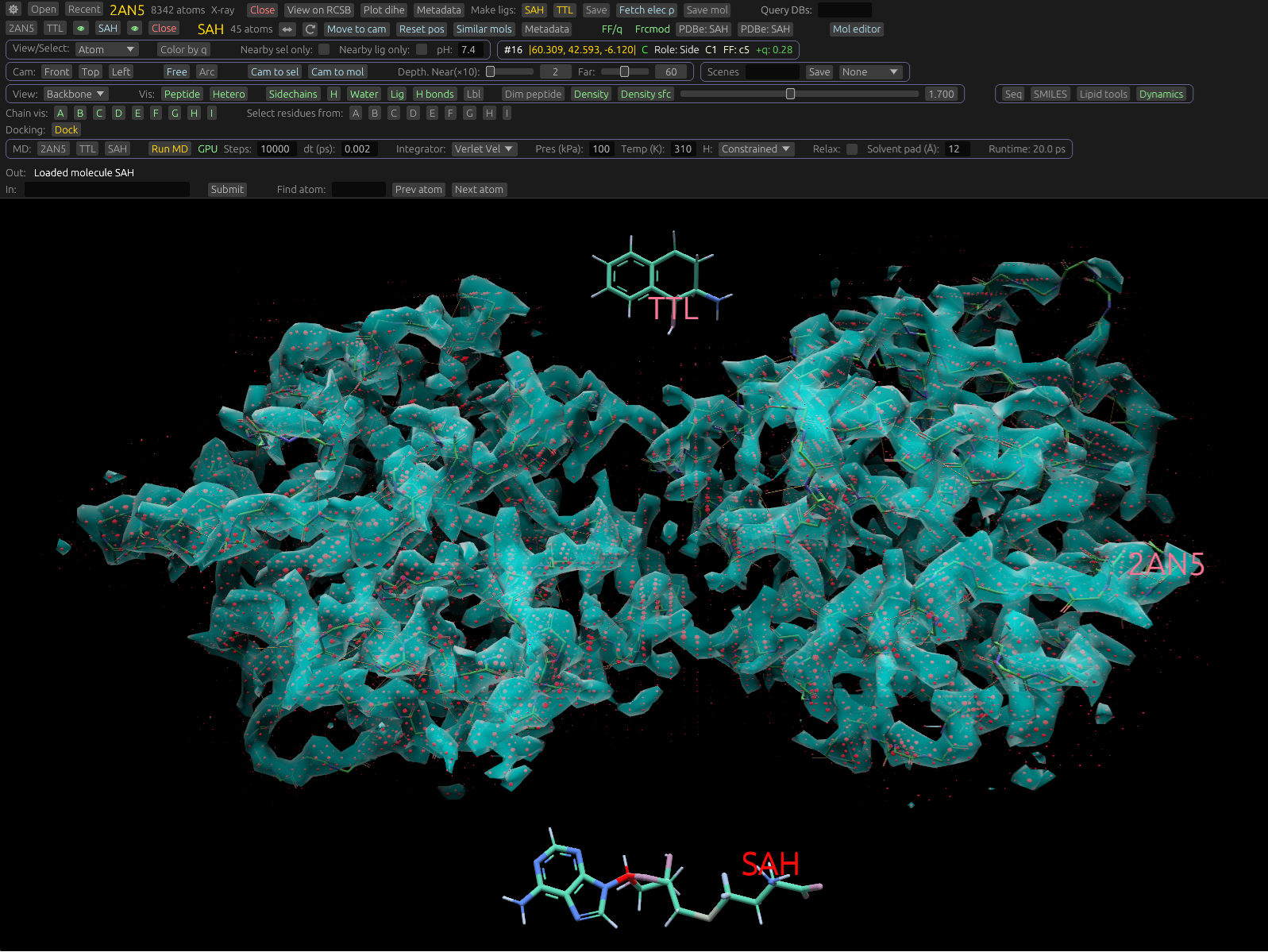
For technical users
File formats
mmCIF, SDF, Mol2, PDBQT, MAP, MTZ. Amber dat/lib/frcmod/prmtop, GROMACS top.
GPU / CPU fallback
CUDA on RTX 3-series+ when present; otherwise multi-core + SIMD.
CLI commands
Subset of PyMOL-style commands for fetch/load/show/edit and camera ops.
Need full instructions? See the README in the repository for compiling, CUDA setup, electron density notes, and hotkeys.