Molchanica
A visual tool for structural biology and drug-design
Load from PDB, PubChem and other databases. Run MD, view electron density, and edit molecules.
Computer-assisted drug design
Perform virtual screening using pharmacophores. Compute ADME and toxicity of any molecule using machine learning. Align molecules.
Compatible
Supports a variety of standard formats for molecules, electron density measurements, MD trajectories/config, and more.
Versatile
Specialized tools for molecular docking, building nucleic acids, and simulating liposomes and lipid nanoparticles (LNPs). Drug-development features including molecule alignment, ADME and toxicity prediction, and pharmacophore-based screening.
Molchanica uses a powerful visualization and camera system that lets you explore molecular systems without friction. Freely navigate your view throughout the system.
ORCA integration for quantum chemistry. GROMACS integration for MD. OpenDDE integration for structure prediction.
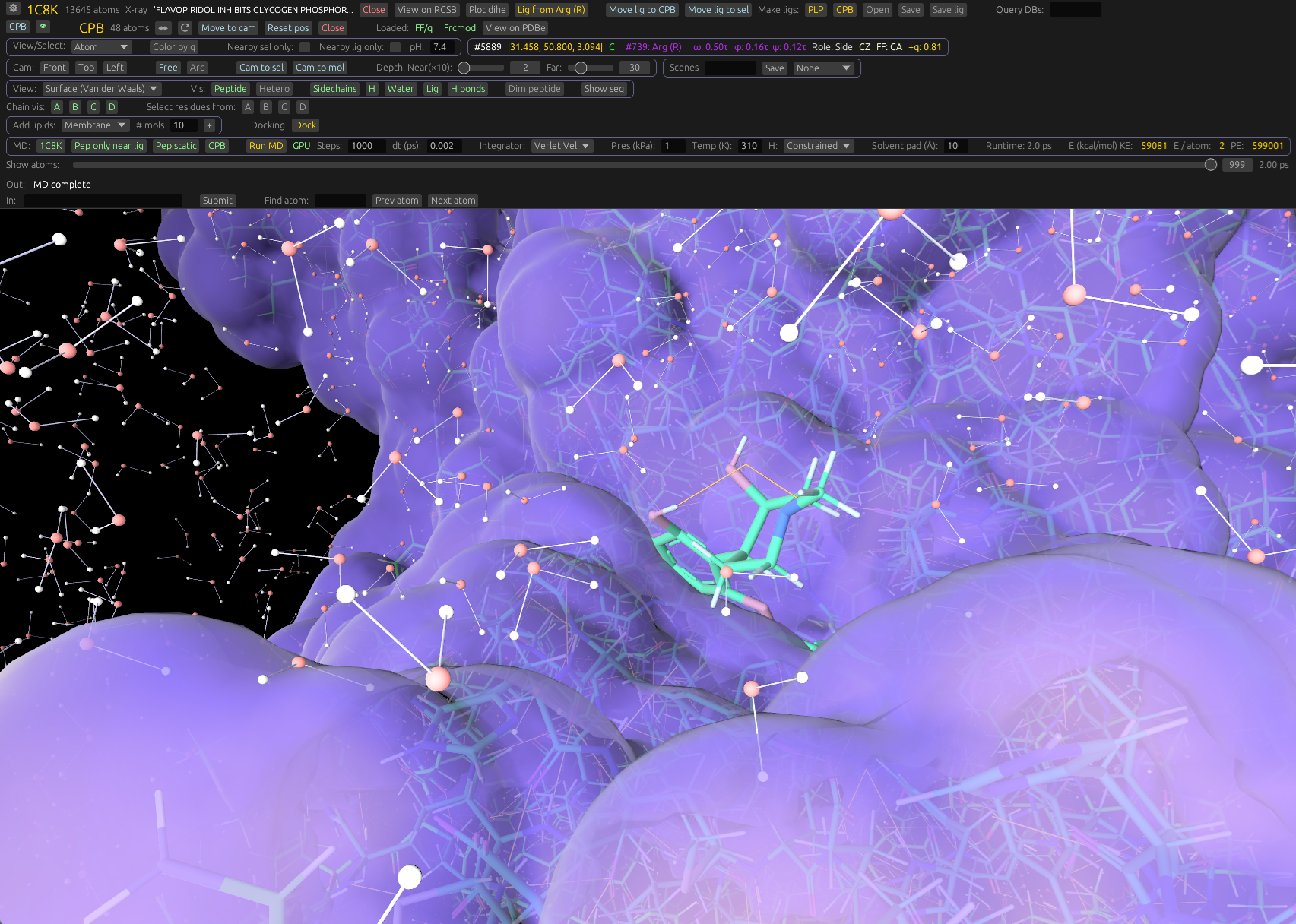
Why molchanica
MD and structure prediction built in
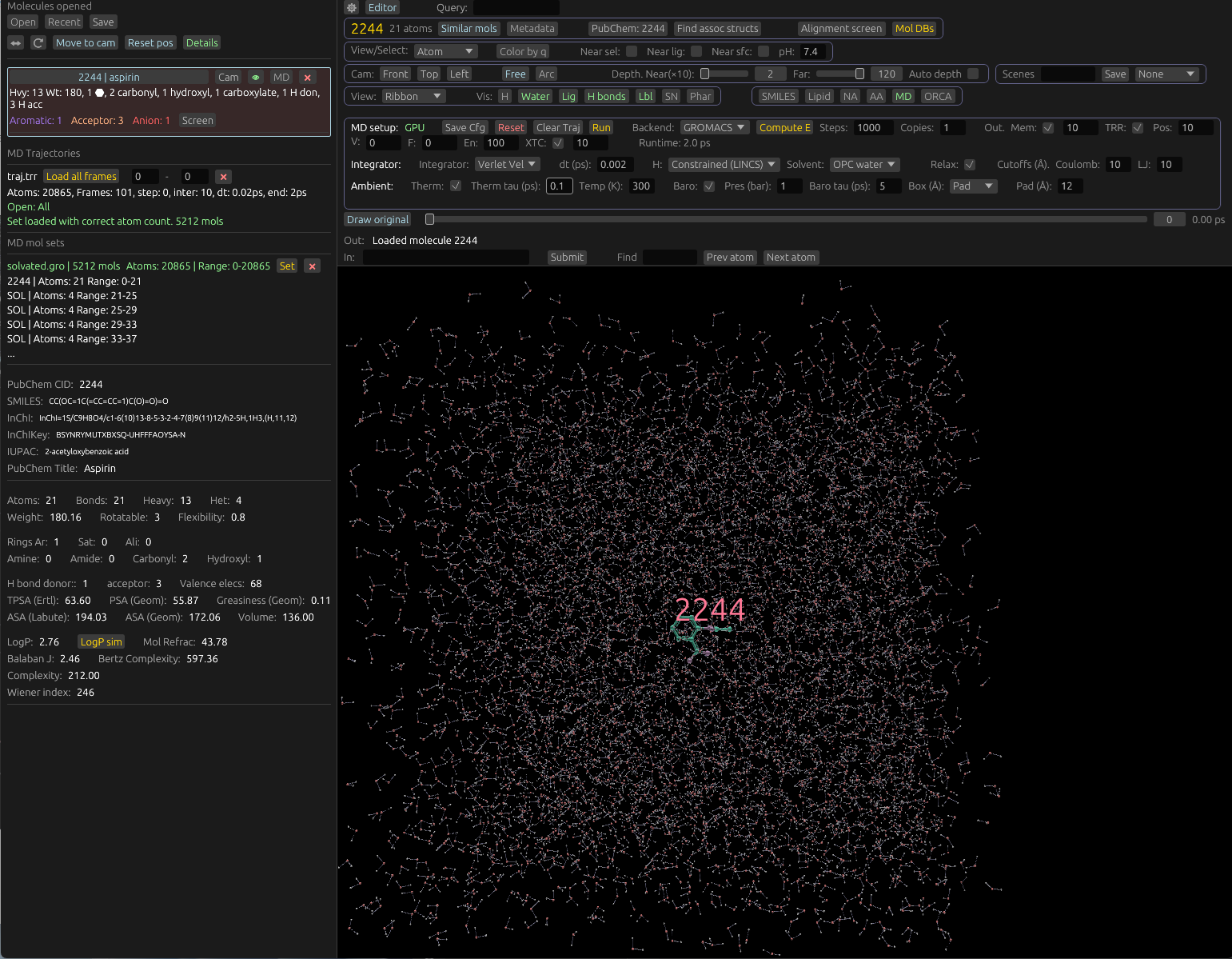
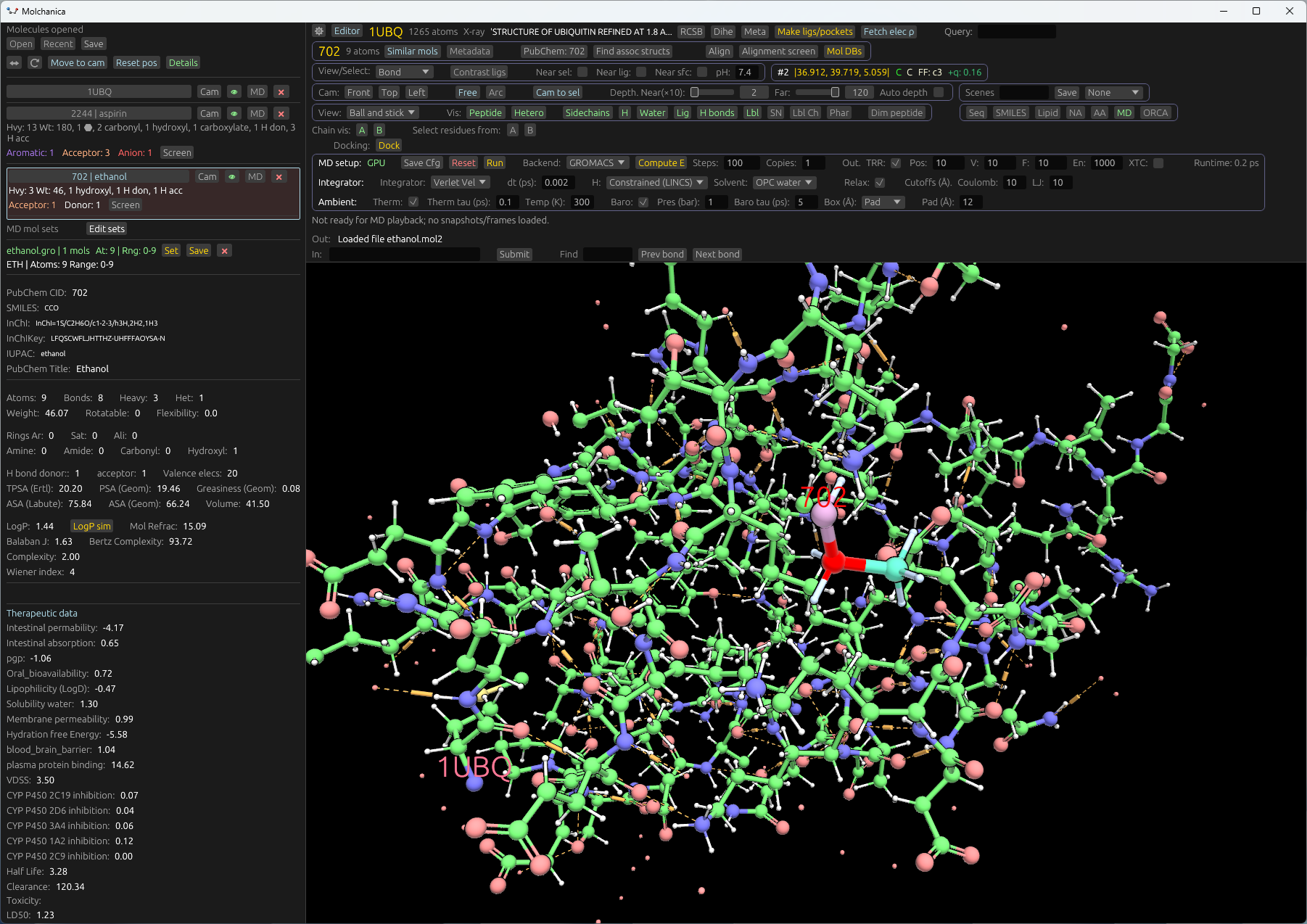
Run molecular dynamics with Amber force fields and OPC water directly in the viewer. Use our own engine, or GROMACS. Compatible with many MD and mol formats. View trajectories loaded from any program.
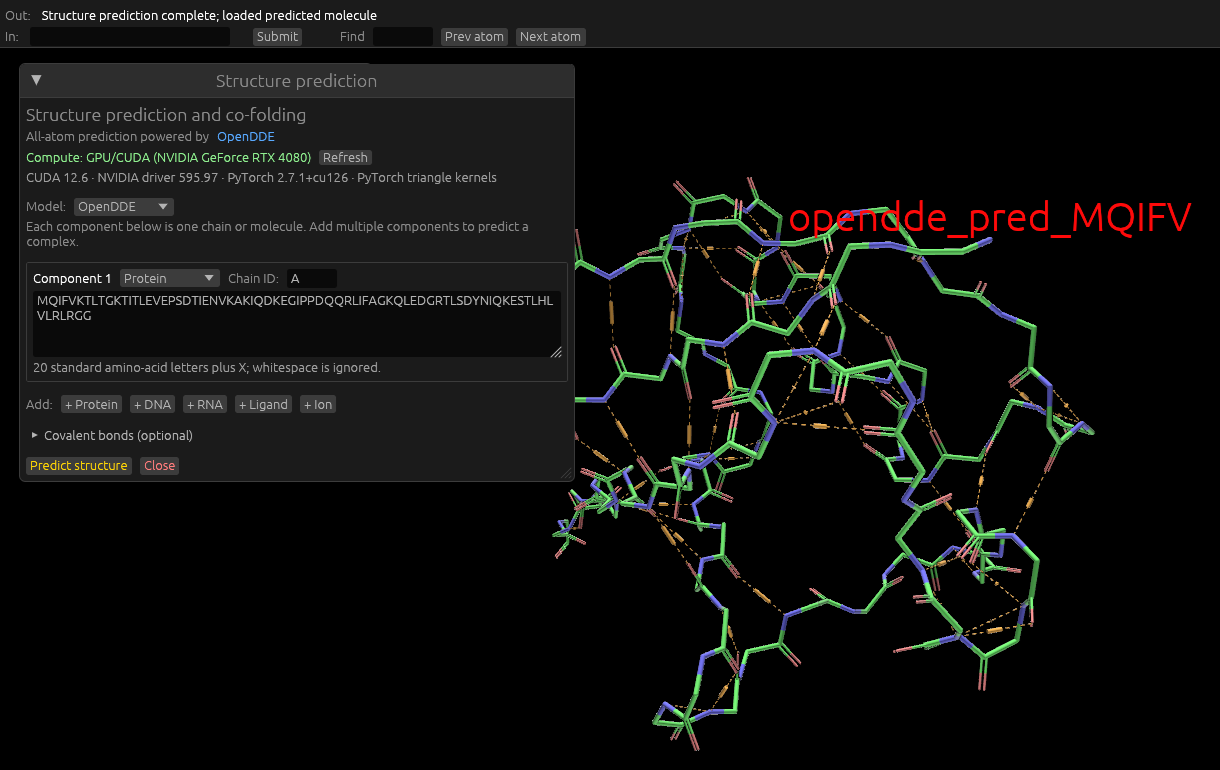
Run OpenDDE natively: A highly accurate machine learning model (Outperforms AlphaFold3) which predicts structure of proteins, small molecules, DNA, RNA, and ions from an input sequence.
Drug development
Molecule screening, alignment, pharmacophores, and ADME using ML.
Density and docking
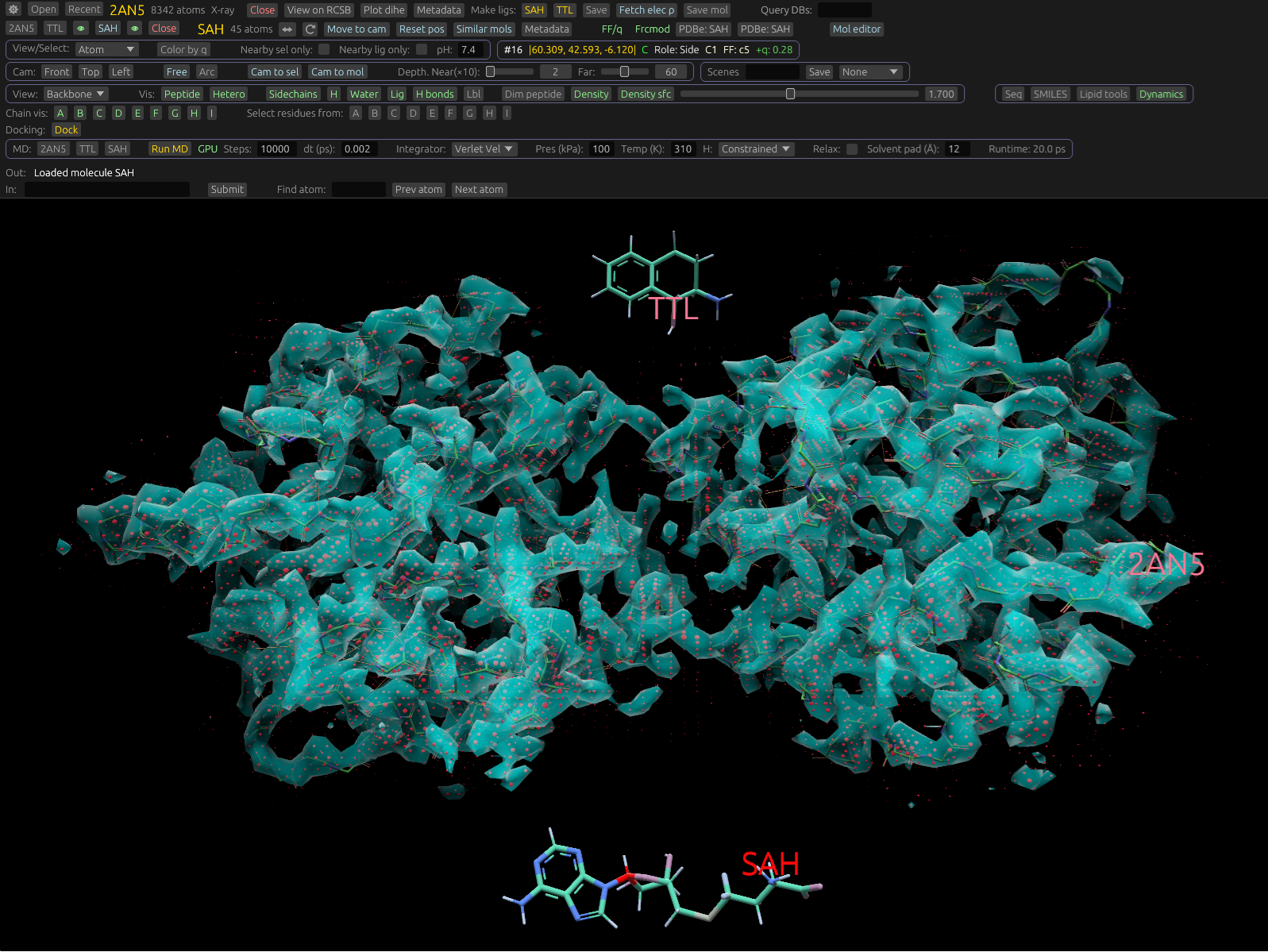
Render 2Fo-Fc maps, isosurfaces, and visualize docked ligands from crystallography or Cryo-EM data.
Fast
Rust, CUDA, SIMD, and thread pools for parallelization. Tight integration with RCSB, PubChem, DrugBank, and PDBe.
Gallery
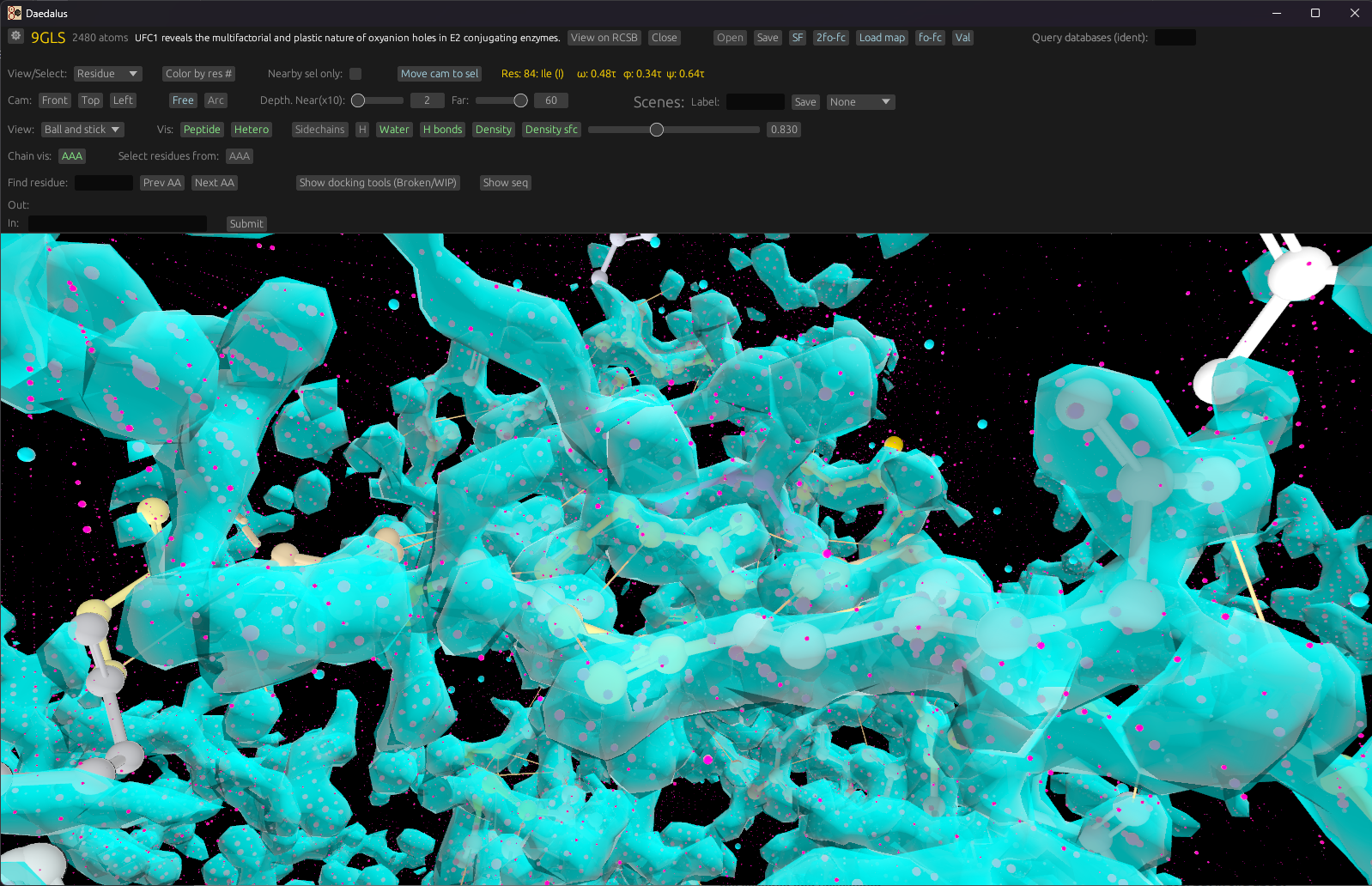
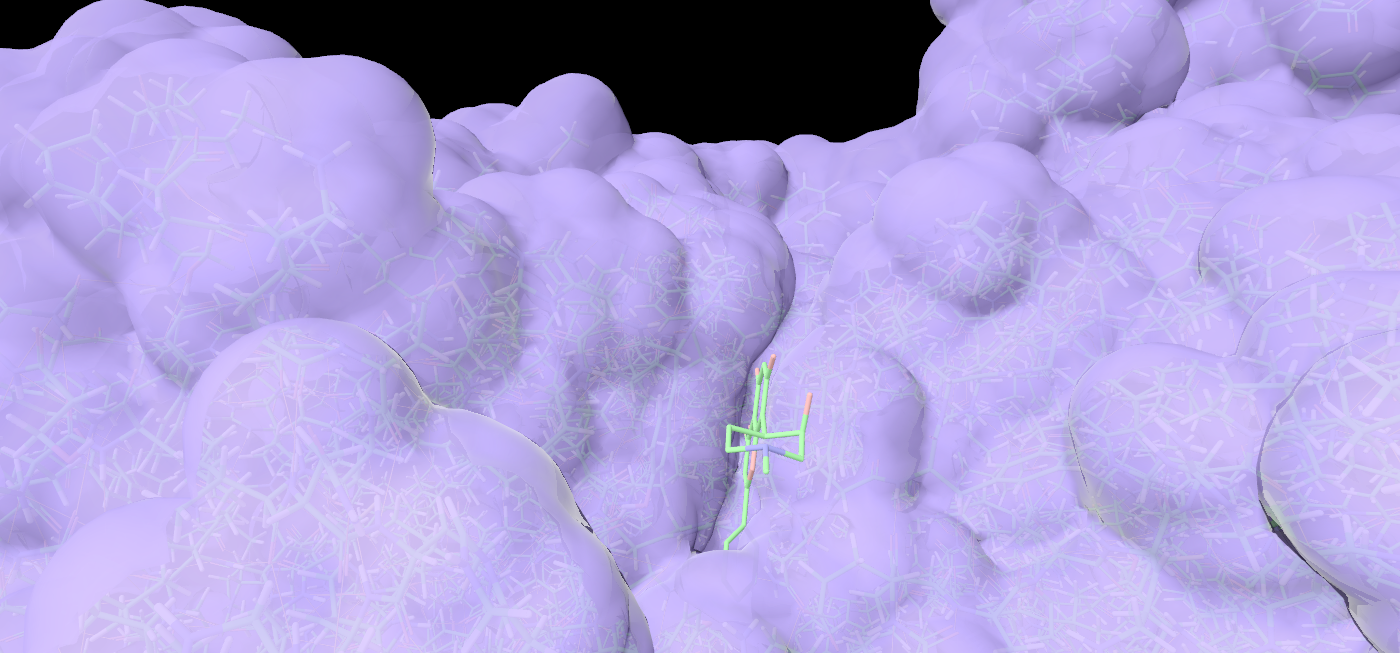
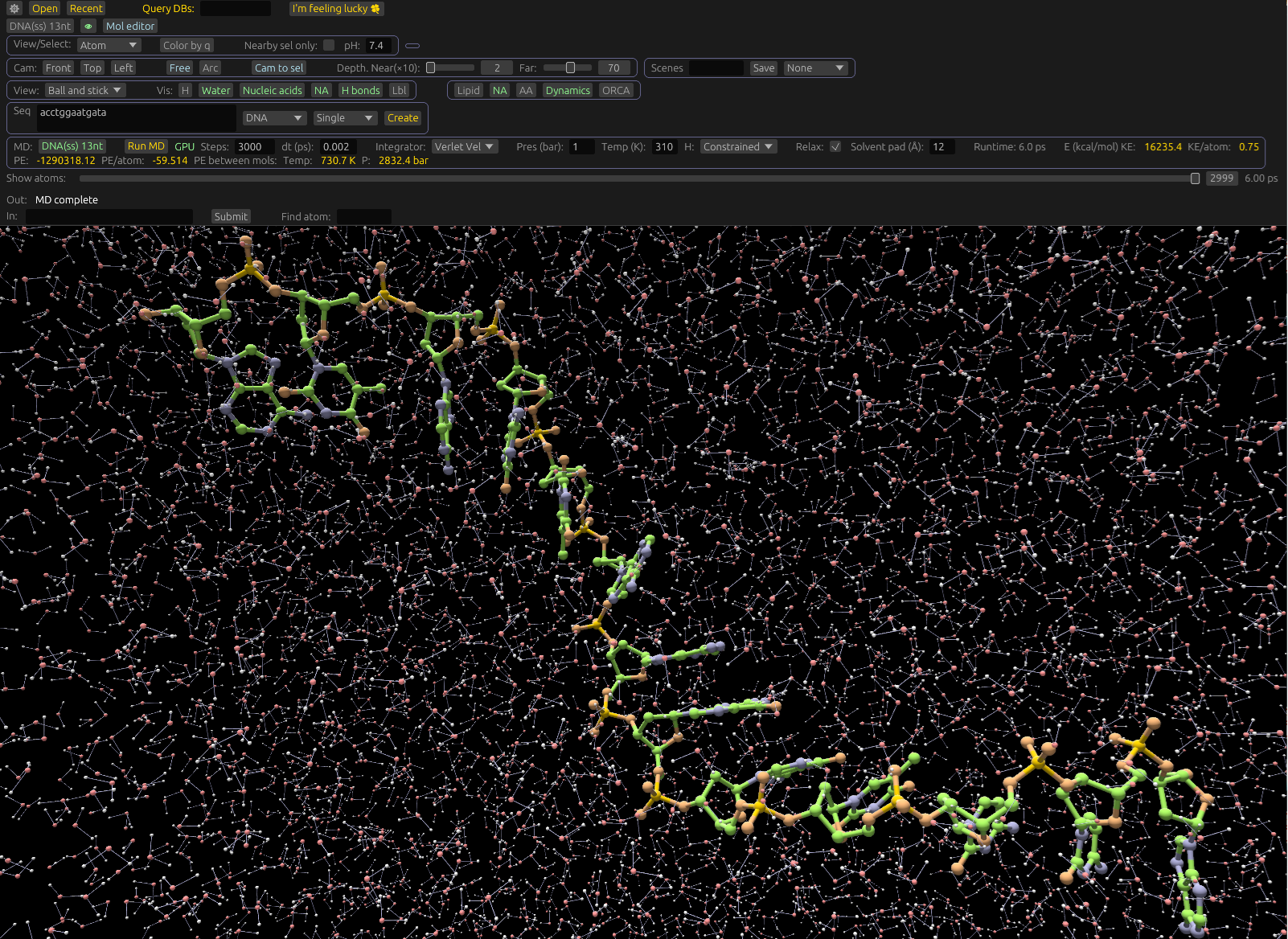
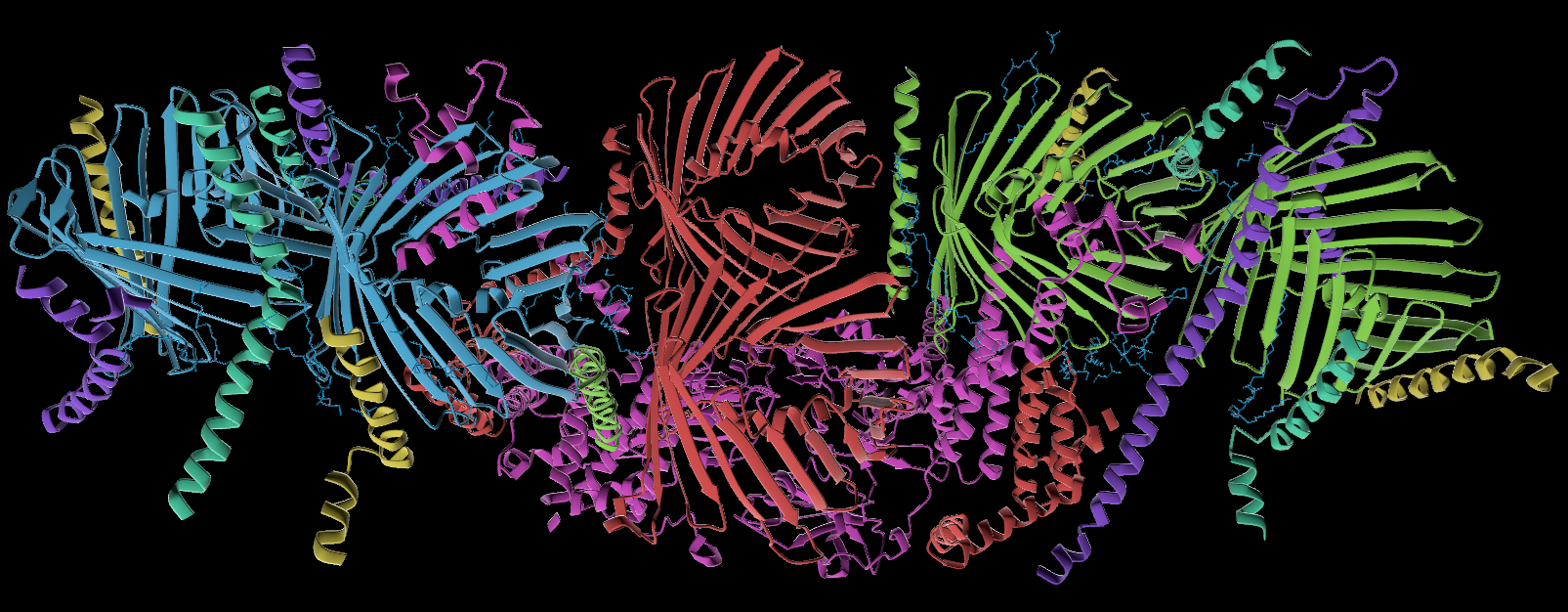
Details
File formats
Supports the most common formats for interop with other software. This includes mmCIF, SDF, Mol2, PDBQT, XYZ, MAP, MTZ, DCD. Amber dat/lib/frcmod/prmtop, GROMACS top.
GPU and CPU parallization
CUDA on Nvidia GPUs; otherwise multi-core + SIMD CPU operations.
CLI commands
Subset of PyMOL-style commands for fetch/load/show/edit and camera ops.
Need full instructions? See the README in the repository for compiling, CUDA setup, electron density notes, and hotkeys.