Molecule editor
This application includes a small-molecule editor. This is 3D only, and allows you to create and change small organic molecules. It's integrated with the molecular dynamics (MD) engine, which it uses to enforce realistic geometry, and demonstrate vibrations. MD can be run continuously, with or without an explicit water solvent. For details on this MD sim, see the associated docs page.
The molecule editor is a separate operating mode from the primary one. It shares the visualization and most hotkeys from elsewhere, but focuses on a single small organic molecule, and has a different user interface. This is a simpler one, which
Currently, this mode only supports the Arc camera. This is appropriate here, as the molecules are expected to be small enough to visualize in their entirely from a single viewpoint. As atoms are added, removed, and moved, the arc enter automatically changes to the molecule's new centroid.
Note that selections in the editor work differently from in the primary mode: Instead of selecting between atoms, bonds, and residues, you can right-click on any atom or bond, and it will select it.
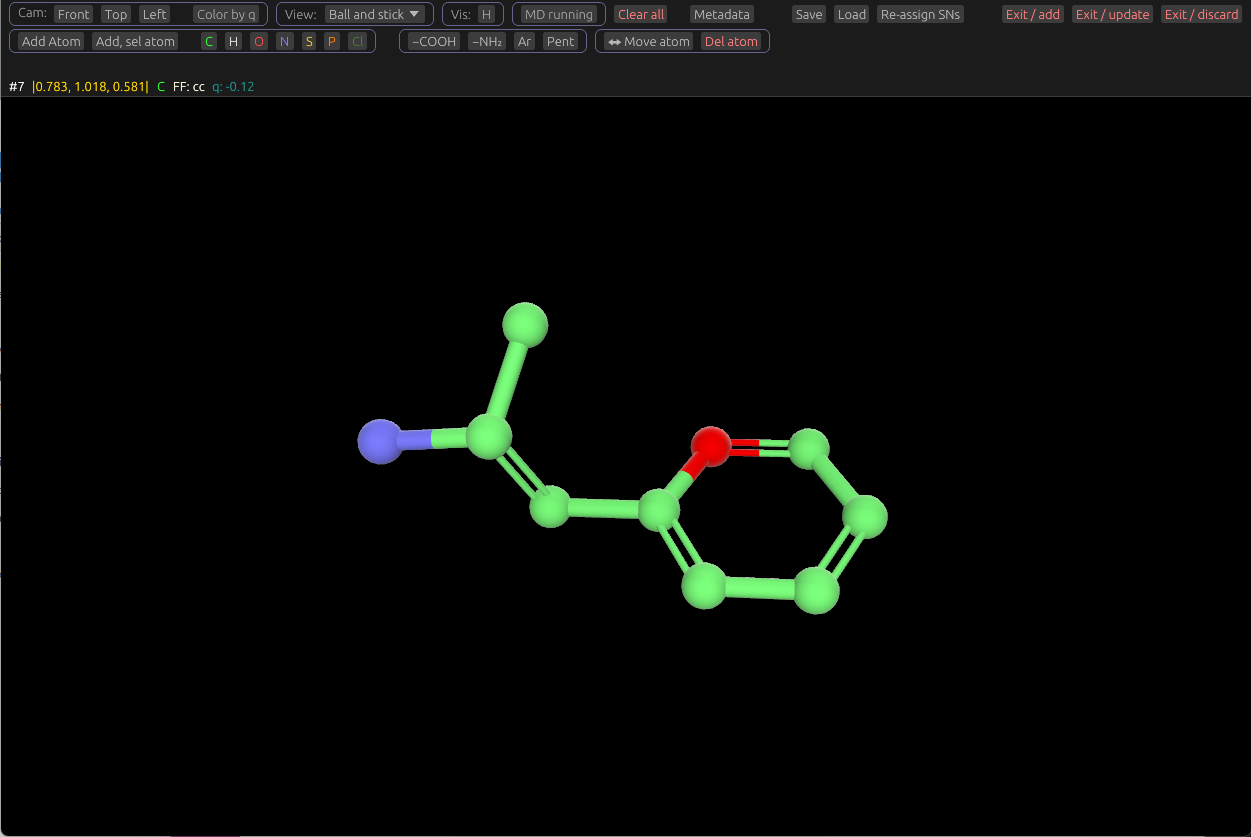
Adding and removing atoms and bonds
To add an atom, first select one, by right-clicking one or more. Then click the Add Atom button in the UI. This will add a new carbon atom covalently bonded to the selected one, with tetrahedral geometry. Hydrogens will re-arranged on the parent atom (The one you selected), and the newly-added one. If multiple atoms are selected, this action will be repeated for each.
You can change this to a different element by selecting it, then clicking one of the color-coded element buttons. For example, click O to turn it into an Oxygen. To change the bond type, select the bond with right-click, then click one of the bond type buttons: - (Single), = (Double), Tr (Triple bond), and Ar (Aromatic). If multiple atoms or bonds are selected, these element and bond-type change buttons will be allied to all selected.
Moving atoms and rotating segments
Moving and rotating shares similarities with the molecule movement and rotating of the primary operating mode, but acts on atoms and bonds, instead of whole molecules. The controls, including how to enter/exit movement/rotation modes, and using the mouse to perform these manipulations are the same. See the manipulation docs section for details. M enters or exits atom movement mode, R enters or exits rotation around a bond, and Esc exits manipulation modes.
In the editor, movement applies to a single atom (the selected one), vs the whole molecule. When in movement mode, the selected atom will change color, indicating it can be moved with the mouse X and Y axes, and the scroll wheel.
Rotation means rotation around a bond. When rotating, one side is considered the anchor, while the other rotates relative to it. The side of the bond with more atoms is always the anchor. When in rotation mode, the bond selected will change color to indicate it's the rotation pivot. Note that you can only rotate around bonds that have free movement, as dictated by geometry. For example, you can't rotate around a bond that's part of a ring. When rotating in the editor, only the mouse horizontal (X) axis controls rotation; moving it up and down, or using the scroll wheel has no effect, unlike in other manipulation modes. The image below demonstrates a molecule in rotation mode, with the bond between two rings as the pivot. Note that the only other bond which can be a pivot in this molecule is the one between the Carbon and Hydroxyl O.
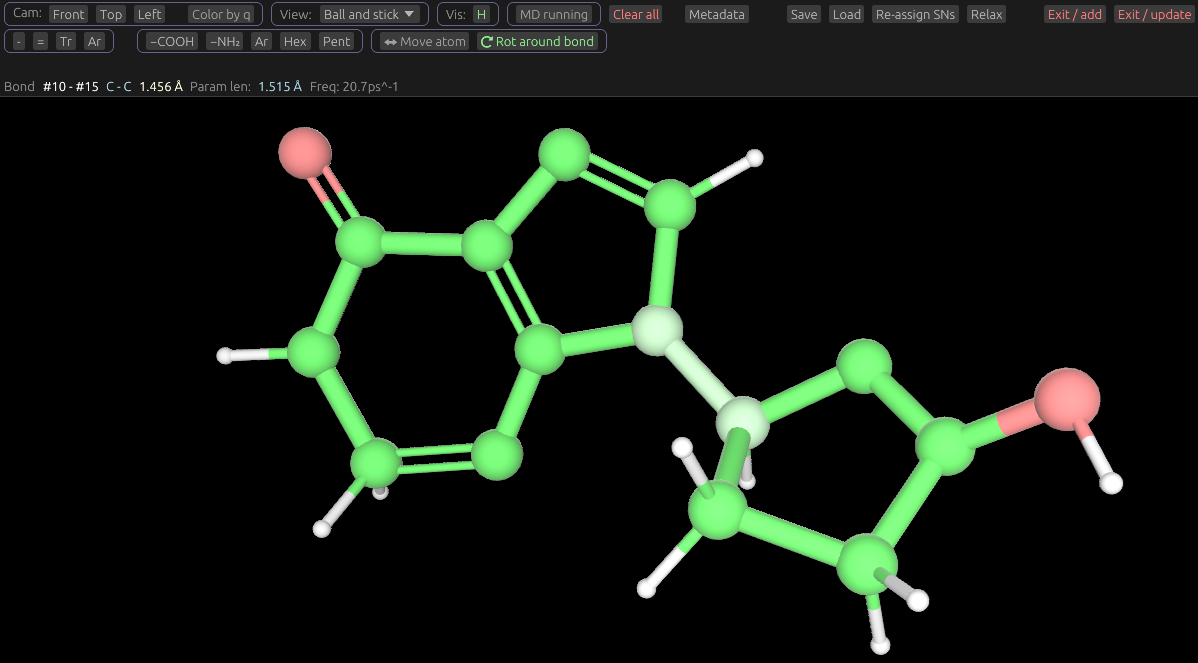
Note that manual manipulations will often be overridden by MD, but these manipulations helps with placing atoms and bonds. As in the primary mode, you can edit movement or rotation by clicking its (Highlighted) UI button, its hotkey (M or R), or using the Esc key.
Relaxing the molecule (Energy minimization)
You can click the Relax button to update the molecule's geometry to be at a local energy minima with regard to its molecular dynamics parameters. For example, this sets up bond lengths and angles correctly if able, regularizes rings, etc. This is done as part of the molecular dynamics engine, using the forces outlined in the MD section of these docs You may wish to run this after adding atoms and bonds to am molecule, moving them, and before saving. You can run this with or without the continuous MD simulation running.
Adding rings and functional groups
To add a ring or functional group, select either an atom or bond, then click one of the appropriate buttons. In the case of rings, if a bond is selected, the ring will be created incorporating this bond, and its two atoms, as part of its structure. You can use this, for example, to create fused rings.
Forcefield parameters and partial charges
Every time you add, remove, or change an atom, partial charges, and Amber force field types are recomputed for the whole molecule. This is relatively quick, (Takes a few ms for most molecules on most computers) and ensures that the dynamics sim stays up to date. You can inspect these by selecting atoms and bonds, and viewing their information in the GUI.
Solvation
The editor uses the same explicit solvation model as the primary mode's MD simulation: OPC rigid 4-point water molecules surrounding the atom. Unlike in the primary mode, you can't view these; they act to realistically interact with and stabilize the molecule you're editing.
Chemistry constraints
(todo)
Adjusting the MD sim
(todo)
Opening and saving
To enter the editor, click the Mol Editor button from the UI. If you open the molecule editor while a small organic molecule is set as active, the editor will open it. If not, it will start with a pair of bonded Carbon atoms
While editing, you can save the molecule directly to a file (e.g. SDF, Mol2) by clicking the Save button. To load the molecule back into the main program, click Exit / add or Exit / update. These add an additional molecule to the primary mode, or replace the one being edited, respectively. Click Exit / discard to return to the primary view without updating anything it. Warning: This will discard all edits you've made since the last time you saved it to disk.
Hydrogen atoms
The editor does not allow you to place or manipulate hydrogen atoms directly: These are managed automatically. They're added, removed, and moved to support realistic geometry. For example, hydrogen atoms are added to each carbon atom until it has 4 bonds total, taking into account double, triple, and aromatic bonds. Hydrogens are repopulated whenever an atom's element changes, or a bond to it changes type or is added or removed.
Tautomers
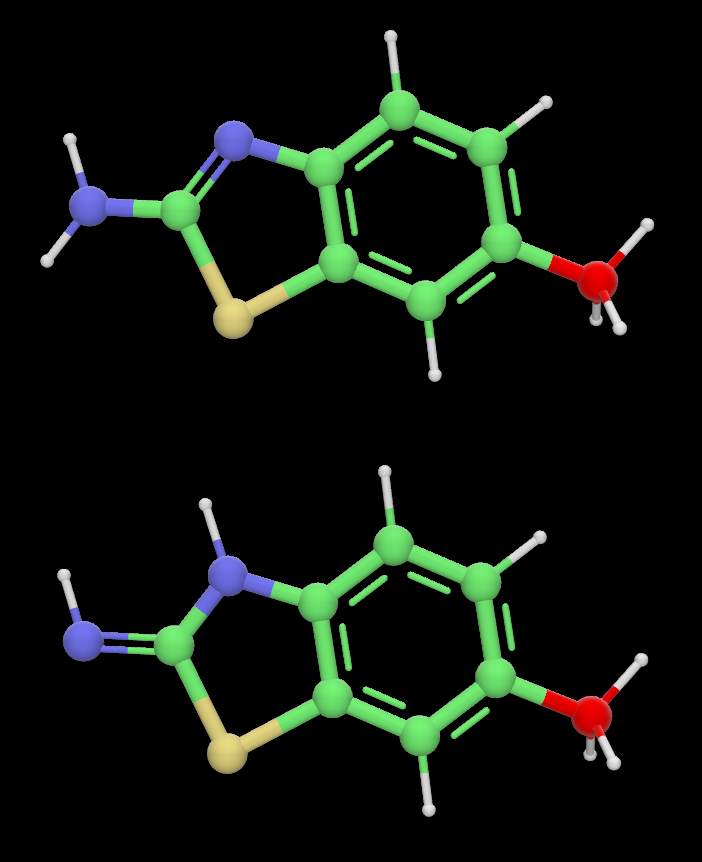
To cycle through Tautomers of the molecule being edited, click the Tautomer button. These are structural isomers that readily interconvert. It shifts around single vs double bonds, and which corresponding atoms have hydrogens bound.
Example of two tautomers below; the Tautomers button toggles between these two configurations: