Opening and saving molecules
Molchanica supports the following file formats:
- Proteins: mmCIF (aka PDBx)
- Small molecules: SDF, Mol2, GRO, XYZ, and PDBQT
- Electron density: 2fo-fc mmCIF, Map, and MTZ
- Force field parameters: dat, lib, frcmod, prmtop (Amber), and top (GROMACS)
Using the sidebar, and opening files
The sidebar, displayed on the left side of the application window, manages opened molecules.
There are several ways open a file: - Click the Open button at the top of the sidebar, and select which molecule to open, filtered by extension. - Drag a file from your operating system's file manager into the application window. - Click the Recent button on the sidebar, then select from recently-opened molecules and other files.
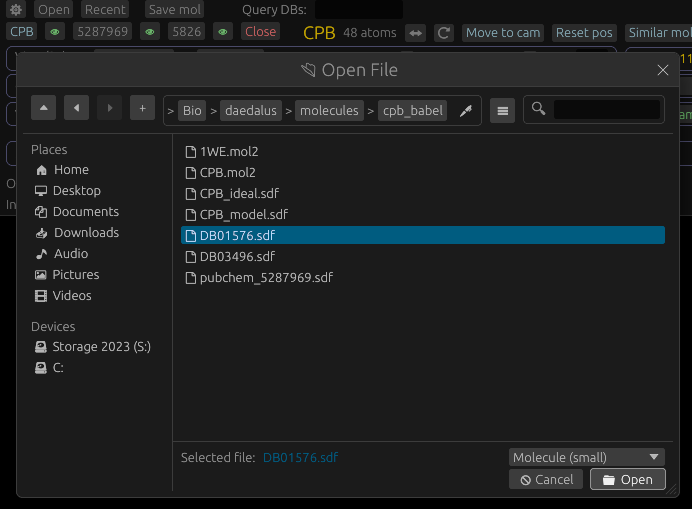
Example of the Recent button:
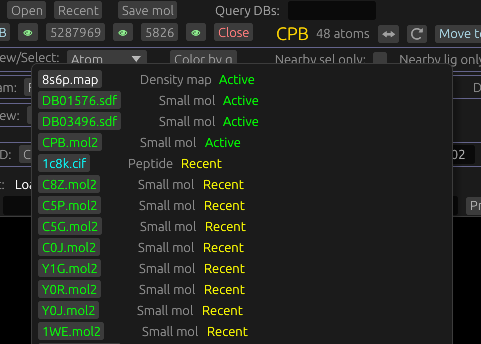
You can also associate file extensions with Molchanica using your operating system's settings, so double-clicking a file in its GUI, etc will open it. Molchanica does not set up file associations on its own; this is up to you.
You will see a row in the sidebar for each opened molecule. In this row, you can click that molecule's button to make it active, or middle-click it to close it. You can click the eye button to toggle visibility, and the X to close it. You can also select or deselect the molecule for an MD sim by clicking MD, or click Cam to move the camera to the molecule.
Loading from SMILES
To create a small molecule from a Simplified Molecular Input Line Entry System (SMILES) string, enter it in the Query box at the top of the UI, then press enter, or click Load from SMILES. Note that the 3D geometry of the loaded molecule may not be exact, but this can be updated in the molecule editor by clicking Relax, to minimize the energy of the geometry. Note that if the SMILES string corresponds to a molecule in PubChem, Molchanica will automatically load PubChem metadata for it.
You may wish to save as a Mol2 or SDF file after loading from SMILES.
Loading molecules from online databases
In addition to loading molecules from the file system, you can load them directly from online databases by dating a molecule's identifier, or common name in the Query DBs field, then pressing enter, or clicking one of the database buttons that appears. See the Datbases section of this documentation for details.
Positioning of opened molecules
Newly opened molecules are placed directly in front of the camera. If there is already a molecule in this position, they're offset as to not overlap, and the camera is moved to look at the newly-opened molecule.
Protein exception
Currently, only one protein can be opened at a time, and its information and controls are located in a dedicated UI section, above that for other molecules. We plan to change this in future versions.
Selecting, closing, and hiding molecules
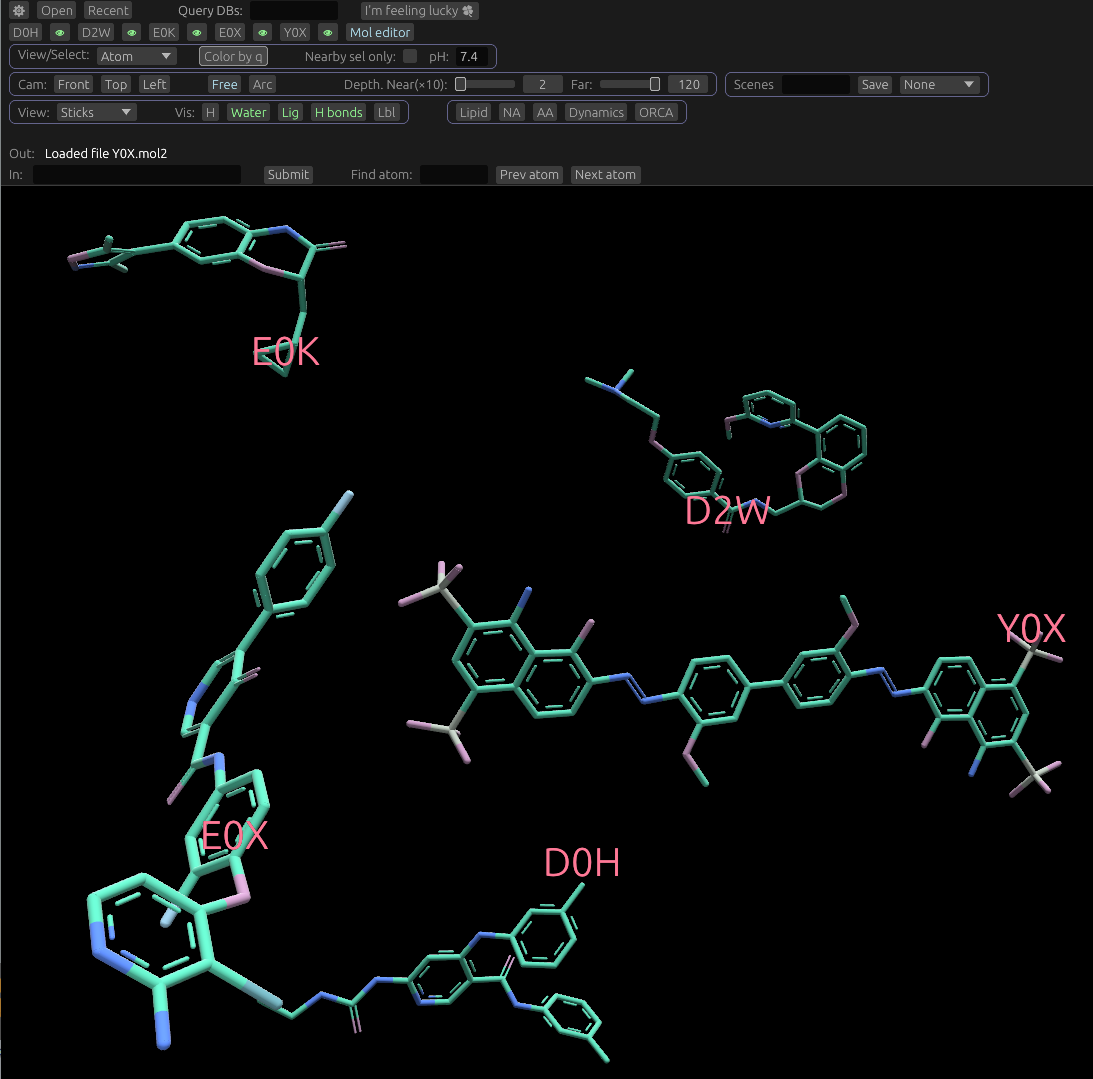
Up to a single molecule can be active at a time. This has a few effects, like determining which molecule to display detailed data for in the UI. To make a molecule active, either select one of its atoms, bonds, or residues (e.g. with right click), or click its identifier in the list of open molecules displayed in the UI, pictured here. Click the eye icon to toggle if this molecule is hidden or not. To close the active molecule, click its Close button in this part of the UI, or middle click its identifier button.