GROMACS integration: Molecular dynamics
Molchanica includes GROMACS integration - you can run MD simulations using it, using the same UI as for our native (dynamics library simluations.) See the Dynamics page of these docs for details on how to use this UI. GROMACS is a powerful, industry standard tool for running molecular dynamics simulations. It has been inspirational for our own dynamics implementation, and configuration. Reference its User Guide for a description of the most important functionality, and its Reference Manual for details about its algorithms.
To use this functionality, you must Install GROMACS on your system. This may involve compiling it from source. It must then be exposed on your system's Path environment varible, and be properly associated with its data directory. The details of this will depend on how GROMACS is comiled; details on this are out of scope of Molchanica, and we do not provide GROMACS executables.
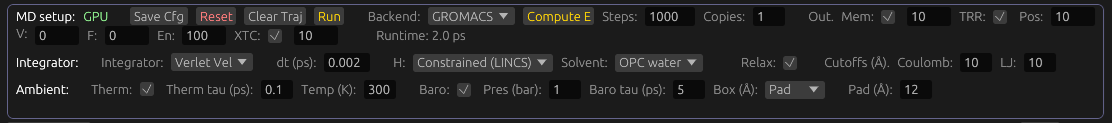
To run, open the MD toolbar, and select GROMACS in the Backend dropdown. If you don't see this dropdown, it means Molchanica was unable to locate GROMACs. On startup, it calls gmx -version, and parses the output. If this command fails, or produces invalid output, the Backend dropdown is hidden, and MD is only avilable using our native (dynamics library) implementation.
We have not implemented all GROMACS options in the GUI, but have included the most common ones. We've ommitted settings which only have one non-deprecated option. If you see something missing you'd like to use, send us an email, and we'll add it.
À la carte
You can mix and match Molchanica GROMACS integration with other tools, as it works by generating and reading standard GROMACS file formats.. For example, you can use Molchanica to generate these files, then run them in a separate workflow. Or use Molchanica to generate and run, then view trajectories in VMD. Or configure and run a simulation using your own scripts, then view the output trajectories and thermodynamics/energy parameters in Molchanica.
For example, you can run MD in any program which can generate a DCD, XTC, or TRR trajectory, then load this directly in Molchanica to view the output molecules at each frame. Or save Molchanica's output from any MD backend, and view it in VMD or another viewer. You can save MD configs as a .mdp file, or load these. Our native force field param structs can be saved in .top fields, and molecules can be saved in .gro format.
For details and saving and loading MD data, see the MD page.
Viewing GROMACS input files
The .mdp data generated, as well as gmx commands (gmx grompp, gmx solvate, and gmx run) are printed to the application log window, and can be viewed on disk.
Solvents
Note: We use the dynamics library to generate solvent boxes with periodic boundary conditions using templates, e.g. for OPC water. We use this to generate .gro files, which we send to gmx solvate. GROMACS uses this template to fill the simulation box. This includes a 60Å cube of water molecules.
Reading GROMACS outputs
After running MD with GROMACS, output data is immediately loaded into to Molchanica. You can also use Molchanica as a viewer for GROMACS output generated elsewhere, e.g. from running GROMACS directly. To do so, load a GROMACS trajectory in .trr, .xtc, or .dcd format into Molchanica using its standard open functionality: e.g. using the Open button, or dragging a file into the window.
You can use the snapshot viewer to navigate between snapshots (aka frames), and view the output in 3D.